小白菊内酯(Parthenolide欧苷菊) NF-κB活化抑制剂/HDAC1抑制剂|CAS 20554-84-1
产品说明书
FAQ
COA
已发表文献
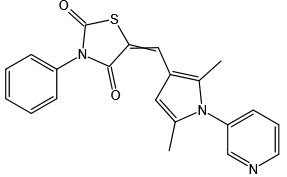
Parthenolide ((-)-Parthenolide),又称小白菊内酯或欧苷菊,是来源于药草短舌匹菊的倍半萜内酯,是NF-κB抑制剂,表现出抗炎、抗肿瘤和抗病毒活性。它传统上主要用来治疗发热、偏头痛、皮肤感染和类风湿性关节炎。Parthenolide还可不依赖NF-κB特异性抑制HDAC1,而不影响其他I/II类HDACs。
产品性质
|
英文别名 (English Synonym) |
Parthenolide, (-)-Parthenolide |
|
中文名称 (Chinese Name) |
小白菊内酯,欧苷菊 |
|
靶点 (Target) |
NF-κB, HDAC1 |
|
通路 (Pathway) |
NF-κB |
|
CAS号 (CAS NO.) |
20554-84-1 |
|
分子式 (Formula) |
C15H20O3 |
|
分子量 (Molecular Weight) |
248.32 |
|
外观 (Appearance) |
粉末 |
|
纯度 (Purity) |
≥98% |
|
溶解性 (Solubility) |
溶于DMSO |
|
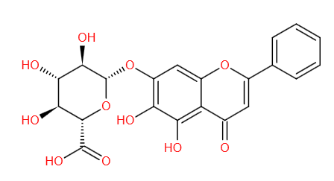
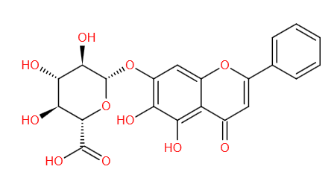
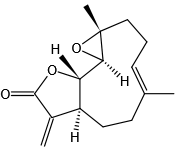
结构式 (Structure) |
|
运输和保存方法
冰袋运输。粉末直接保存于-20℃,有效期3年。建议分装后-20℃干燥保存,避免反复冻融。
注意事项
1. 为了您的安全和健康,请穿实验服并戴一次性手套操作。
2. 粉末溶解前请先短暂离心,以保证产品全在管底。
3. 请勿吸入、吞咽或者直接接触皮肤和眼睛。
4. 本产品仅用于科研用途,禁止用于人身上。
使用浓度
【具体使用浓度请参考相关文献,并根据自身实验条件(如实验目的,细胞种类,培养特性等)进行摸索和优化。】
使用方法(数据来自于公开发表的文献,仅供参考)
(一)细胞实验(体外实验)
小白菊内酯以浓度和时间依赖方式诱导肺癌细胞(Calu-1、H1792、A549、H1299、H157和H460)凋亡。小白菊内酯在A549细胞中以浓度依赖性方式诱导G0/G1细胞周期停滞,在H1792细胞中诱导G2/M细胞周期停滞。[1]
(二)动物实验(体内实验)
在胆管结扎的Phb1 KO小鼠模型中,腹腔注射小白菊内酯(3 mg/kg),降低了小鼠的死亡率,减少了胆管结扎后的肝脏损伤,延迟了肝纤维化。[2]
参考文献
- Zhao X, et al. Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J Exp Clin Cancer Res. 2014 Jan 6;33:3.
- Barbier-Torres L, et al. Histone deacetylase 4 promotes cholestatic liver injury in the absence of prohibitin-1. Hepatology. 2015 Oct;62(4):1237-48.
- Nakshatri H, et al. NF-κB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis. 2015 Jan 22;6:e1608.
HB221114
Parthenolide ((-)-Parthenolide),又称小白菊内酯或欧苷菊,是来源于药草短舌匹菊的倍半萜内酯,是NF-κB抑制剂,表现出抗炎、抗肿瘤和抗病毒活性。它传统上主要用来治疗发热、偏头痛、皮肤感染和类风湿性关节炎。Parthenolide还可不依赖NF-κB特异性抑制HDAC1,而不影响其他I/II类HDACs。
产品性质
|
英文别名 (English Synonym) |
Parthenolide, (-)-Parthenolide |
|
中文名称 (Chinese Name) |
小白菊内酯,欧苷菊 |
|
靶点 (Target) |
NF-κB, HDAC1 |
|
通路 (Pathway) |
NF-κB |
|
CAS号 (CAS NO.) |
20554-84-1 |
|
分子式 (Formula) |
C15H20O3 |
|
分子量 (Molecular Weight) |
248.32 |
|
外观 (Appearance) |
粉末 |
|
纯度 (Purity) |
≥98% |
|
溶解性 (Solubility) |
溶于DMSO |
|
结构式 (Structure) |
|
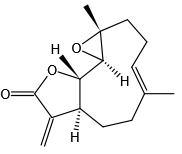
运输和保存方法
冰袋运输。粉末直接保存于-20℃,有效期3年。建议分装后-20℃干燥保存,避免反复冻融。
注意事项
1. 为了您的安全和健康,请穿实验服并戴一次性手套操作。
2. 粉末溶解前请先短暂离心,以保证产品全在管底。
3. 请勿吸入、吞咽或者直接接触皮肤和眼睛。
4. 本产品仅用于科研用途,禁止用于人身上。
使用浓度
【具体使用浓度请参考相关文献,并根据自身实验条件(如实验目的,细胞种类,培养特性等)进行摸索和优化。】
使用方法(数据来自于公开发表的文献,仅供参考)
(一)细胞实验(体外实验)
小白菊内酯以浓度和时间依赖方式诱导肺癌细胞(Calu-1、H1792、A549、H1299、H157和H460)凋亡。小白菊内酯在A549细胞中以浓度依赖性方式诱导G0/G1细胞周期停滞,在H1792细胞中诱导G2/M细胞周期停滞。[1]
(二)动物实验(体内实验)
在胆管结扎的Phb1 KO小鼠模型中,腹腔注射小白菊内酯(3 mg/kg),降低了小鼠的死亡率,减少了胆管结扎后的肝脏损伤,延迟了肝纤维化。[2]
参考文献
- Zhao X, et al. Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J Exp Clin Cancer Res. 2014 Jan 6;33:3.
- Barbier-Torres L, et al. Histone deacetylase 4 promotes cholestatic liver injury in the absence of prohibitin-1. Hepatology. 2015 Oct;62(4):1237-48.
- Nakshatri H, et al. NF-κB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis. 2015 Jan 22;6:e1608.
HB221114

暂无内容
暂无内容